
Projects
Project 1: High-order assembly of MegaTrans complexes for hormone-independent enhancer activation
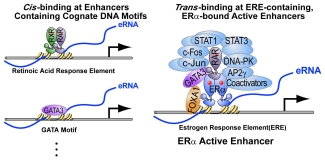 Endocrine therapy is commonly used in hormone-driven breast and prostate cancers. A persistent challenge is disease progression caused by hormone resistance during the treatment. Studies for the past 25 years have revealed an essential role of hormones (i.e., estrogen and androgen) and their receptors, ERα and AR, in cancer progression. Increasing evidence indicates that epigenetic deregulation of ERα/AR-bound enhancers profoundly alters hormone-mediated transcription machinery, leading to the development of hormone resistance. However, the molecular mechanisms underlying this hormone-resistance transition of enhancer function are largely unknown. We have recently discovered that the most active and functionally important ERα-bound enhancers can recruit a large number of DNA-binding transcription factors through protein-protein interactions. These newly identified ERα ‘co-activators’, termed MegaTrans transcription factors (TFs), are required to activate ERα-bound enhancers and also serve as a signature of functional enhancers. Our preliminary data additionally show the presence of MegaTrans TFs in AR-bound enhancers. Because most MegaTrans TFs are signaling-dependent molecules, they may receive other signals from tumor microenvironments to alter enhancer functions. Thus, combinatorial interactions between ERα/AR and MegaTrans TFs make their enhancers respond not only to estrogen or androgen but also to other microenvironmental signals. We hypothesize that the composition and interaction of MegaTrans TFs undergo dynamic changes during cancer progression, resulting in alterations of ERα/AR enhancer functions that promote hormone-resistance in breast and prostate cancer cells.
Endocrine therapy is commonly used in hormone-driven breast and prostate cancers. A persistent challenge is disease progression caused by hormone resistance during the treatment. Studies for the past 25 years have revealed an essential role of hormones (i.e., estrogen and androgen) and their receptors, ERα and AR, in cancer progression. Increasing evidence indicates that epigenetic deregulation of ERα/AR-bound enhancers profoundly alters hormone-mediated transcription machinery, leading to the development of hormone resistance. However, the molecular mechanisms underlying this hormone-resistance transition of enhancer function are largely unknown. We have recently discovered that the most active and functionally important ERα-bound enhancers can recruit a large number of DNA-binding transcription factors through protein-protein interactions. These newly identified ERα ‘co-activators’, termed MegaTrans transcription factors (TFs), are required to activate ERα-bound enhancers and also serve as a signature of functional enhancers. Our preliminary data additionally show the presence of MegaTrans TFs in AR-bound enhancers. Because most MegaTrans TFs are signaling-dependent molecules, they may receive other signals from tumor microenvironments to alter enhancer functions. Thus, combinatorial interactions between ERα/AR and MegaTrans TFs make their enhancers respond not only to estrogen or androgen but also to other microenvironmental signals. We hypothesize that the composition and interaction of MegaTrans TFs undergo dynamic changes during cancer progression, resulting in alterations of ERα/AR enhancer functions that promote hormone-resistance in breast and prostate cancer cells.
Project 2: Fine-scale nucleosome repositioning of enhancers for hormone-independent genomic function
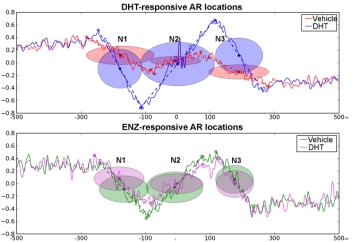 The cognate receptors AR and ERα can remain active for tumor progression after anti-hormone treatment for patients with prostate and breast cancers. Despite intensive efforts to elucidate the underlying mechanisms, little information is available concerning AR/ERα genomic function for promoting hormone resistance at the nucleosome level. In preliminary studies, we observed this genomic function is well-orchestrated, relying on precise nucleosome organization within cis-bound enhancers for hormone-dependent transcription. Interestingly, we also found that this epigenetic mechanism can be hijacked by hormone-resistant cells to gain their growth and invasion advantages. Therefore, we hypothesize that altered nucleosome positions, or nucleosome repositioning, in and near AR/ERα-bound enhancers are being exploited for hormone-independent genomic function in advanced cancers. In Aim 1, we will conduct ChIP-ePENS and MNase-seq to comprehensively map nucleosome boundaries of AR/ERα-bound enhancers in a panel of hormone-sensitive and -resistant cancer cells. RNA-seq will be conducted to determine differential expression patterns of corresponding genes in these cell lines. The NucPat computational pipeline will be deployed to seamlessly process complex omics-seq data (Aim 2). We will use a Kernel Density Estimation algorithm to determine nucleosome positioning and spacing when AR or ERα establishes direct contact with its binding motif. Using a Hidden Markov model, we will identify active nucleosome states that maximize DNA-protein contact for AR/ERα genomic functions. In addition, pioneer factor FOXA1 and chromatin remodelers participate in this nucleosome repositioning even in the absence of agonists or in the presence of antagonists. To confirm this computational modeling in vivo, ChIP-ePENS and MNase-seq will be conducted in patient-derived xenograft (PDX) lines carrying hormone-sensitive and -resistant tumors (Aim 3). A nucleosome-phasing index (NPI) will be established to quantitatively assess the nucleosome states of AR/ERα redeployment in different PDX lines. This integrative omics analysis will be extended to a cohort of primary tumors, categorized into high- and low-risk groups. Again, we will calculate individual NPIs and correlate the data with clinicopathological features of patients. This translational study is intended to determine whether nucleosome phasing for AR/ERα redeployment is already present in high-risk primary tumors. Patients with this intrinsic phenotype are expected to have an adverse clinical outcome, irrespective of their anti-hormone treatments. Therefore, our proposed study will address a previously uncharacterized mechanism of hormone resistance and provide experimental evidence that nucleosome repositioning plays an integral role in redefining AR/ERα genomic function for advanced development of prostate and breast cancers.
The cognate receptors AR and ERα can remain active for tumor progression after anti-hormone treatment for patients with prostate and breast cancers. Despite intensive efforts to elucidate the underlying mechanisms, little information is available concerning AR/ERα genomic function for promoting hormone resistance at the nucleosome level. In preliminary studies, we observed this genomic function is well-orchestrated, relying on precise nucleosome organization within cis-bound enhancers for hormone-dependent transcription. Interestingly, we also found that this epigenetic mechanism can be hijacked by hormone-resistant cells to gain their growth and invasion advantages. Therefore, we hypothesize that altered nucleosome positions, or nucleosome repositioning, in and near AR/ERα-bound enhancers are being exploited for hormone-independent genomic function in advanced cancers. In Aim 1, we will conduct ChIP-ePENS and MNase-seq to comprehensively map nucleosome boundaries of AR/ERα-bound enhancers in a panel of hormone-sensitive and -resistant cancer cells. RNA-seq will be conducted to determine differential expression patterns of corresponding genes in these cell lines. The NucPat computational pipeline will be deployed to seamlessly process complex omics-seq data (Aim 2). We will use a Kernel Density Estimation algorithm to determine nucleosome positioning and spacing when AR or ERα establishes direct contact with its binding motif. Using a Hidden Markov model, we will identify active nucleosome states that maximize DNA-protein contact for AR/ERα genomic functions. In addition, pioneer factor FOXA1 and chromatin remodelers participate in this nucleosome repositioning even in the absence of agonists or in the presence of antagonists. To confirm this computational modeling in vivo, ChIP-ePENS and MNase-seq will be conducted in patient-derived xenograft (PDX) lines carrying hormone-sensitive and -resistant tumors (Aim 3). A nucleosome-phasing index (NPI) will be established to quantitatively assess the nucleosome states of AR/ERα redeployment in different PDX lines. This integrative omics analysis will be extended to a cohort of primary tumors, categorized into high- and low-risk groups. Again, we will calculate individual NPIs and correlate the data with clinicopathological features of patients. This translational study is intended to determine whether nucleosome phasing for AR/ERα redeployment is already present in high-risk primary tumors. Patients with this intrinsic phenotype are expected to have an adverse clinical outcome, irrespective of their anti-hormone treatments. Therefore, our proposed study will address a previously uncharacterized mechanism of hormone resistance and provide experimental evidence that nucleosome repositioning plays an integral role in redefining AR/ERα genomic function for advanced development of prostate and breast cancers.
Project 3: Topological mapping of chromatin architectures for hormone-independent gene transcription
 Long-range chromatin interactions between ERα/AR-bound enhancers and promoters are necessary for coordinated gene regulation in breast and prostate cancer cells. These interactions occur via the formation of 3D chromatin architecture that brings enhancers and transcription factor complexes into close contact with target genes. To decode this complex regulation, we and other investigators have previously used Hi-C to map topologically associated domains (TADs) in different cell types. In a further study, we have identified a cancer-specific TAD on chromosome 17q23 that can be partitioned into an ERα-regulated transcription hub. Concordant up-regulation of its target genes is found to be associated with short disease-free survival in a subgroup of ERα-positive breast cancer patients, irrespective of their anti-hormone treatments. Emerging evidence has also shown AR-specific TADs are present in the prostate cancer cell genome. Therefore, we hypothesize that 1) frequent hormone (i.e., estrogen or androgen) stimulation leads to the formation of ERα/AR-related TADs that dynamically regulate transcription of multiple genes for aberrant proliferation of breast and prostate cancer cells and 2) in the presence of antagonists, a subset of these chromatin domains, herein termed transition TADs, continue to be exploited through chromatin redeployment for hormone-independent transcription. Whereas the majority of ERα/AR-related TADs are functionally suppressed by antagonists, transition TADs may partially escape this blockade for constitutive regulation of gene transcription. To test these hypotheses, we will use a modified Hi-C method, called tethered conformation capture (TCC), to investigate dynamic changes of TAD structures in hormone-sensitive and -resistant cancer cell lines exposed to agonists or antagonists (Aim 1). ChIP-seq of repressive, active, and gene-body histone marks and CTCF insulator will also be conducted in this cell line panel. MNase-seq and MBDCap-seq datasets will be acquired to map euchromatinized and heterochromatinized TADs. To integrate omics-seq data, we will develop a computational model, PRAM3D, which applies a Poisson Random effect Architecture Model (PRAM) to recapitulate 3D chromatin architectures (Aim 2). A Bayesian hierarchical model will predict putative transition TADs that concordantly regulate hormone-independent transcription of target genes. Furthermore, we will use a nucleosome density method to classify transition TAD subdomains into different regulatory categories, i.e., active, repressive, or bivalent transcription hubs. CRISPR/Cas9 genome-editing of critical chromatin regions may functionally disassemble spatiotemporal organization of these TAD-associated hubs (Aim 3). Proliferation and invasion/migration assays will determine whether this genome editing partially re-sensitizes cancer cells to anti-hormone treatments. We will also interrogate the mechanistic contribution of histone modifications and other epigenetic modulators for the establishment of transition TAD structures. In silico expression profiling and single-cell RNA seq will be conducted in primary tumors of TCGA cohorts and in cancer cell subpopulations, respectively, and determine whether concordant regulation of TAD-associated target genes is intrinsic predictors of hormone resistance in the breast or prostate cancer.
Long-range chromatin interactions between ERα/AR-bound enhancers and promoters are necessary for coordinated gene regulation in breast and prostate cancer cells. These interactions occur via the formation of 3D chromatin architecture that brings enhancers and transcription factor complexes into close contact with target genes. To decode this complex regulation, we and other investigators have previously used Hi-C to map topologically associated domains (TADs) in different cell types. In a further study, we have identified a cancer-specific TAD on chromosome 17q23 that can be partitioned into an ERα-regulated transcription hub. Concordant up-regulation of its target genes is found to be associated with short disease-free survival in a subgroup of ERα-positive breast cancer patients, irrespective of their anti-hormone treatments. Emerging evidence has also shown AR-specific TADs are present in the prostate cancer cell genome. Therefore, we hypothesize that 1) frequent hormone (i.e., estrogen or androgen) stimulation leads to the formation of ERα/AR-related TADs that dynamically regulate transcription of multiple genes for aberrant proliferation of breast and prostate cancer cells and 2) in the presence of antagonists, a subset of these chromatin domains, herein termed transition TADs, continue to be exploited through chromatin redeployment for hormone-independent transcription. Whereas the majority of ERα/AR-related TADs are functionally suppressed by antagonists, transition TADs may partially escape this blockade for constitutive regulation of gene transcription. To test these hypotheses, we will use a modified Hi-C method, called tethered conformation capture (TCC), to investigate dynamic changes of TAD structures in hormone-sensitive and -resistant cancer cell lines exposed to agonists or antagonists (Aim 1). ChIP-seq of repressive, active, and gene-body histone marks and CTCF insulator will also be conducted in this cell line panel. MNase-seq and MBDCap-seq datasets will be acquired to map euchromatinized and heterochromatinized TADs. To integrate omics-seq data, we will develop a computational model, PRAM3D, which applies a Poisson Random effect Architecture Model (PRAM) to recapitulate 3D chromatin architectures (Aim 2). A Bayesian hierarchical model will predict putative transition TADs that concordantly regulate hormone-independent transcription of target genes. Furthermore, we will use a nucleosome density method to classify transition TAD subdomains into different regulatory categories, i.e., active, repressive, or bivalent transcription hubs. CRISPR/Cas9 genome-editing of critical chromatin regions may functionally disassemble spatiotemporal organization of these TAD-associated hubs (Aim 3). Proliferation and invasion/migration assays will determine whether this genome editing partially re-sensitizes cancer cells to anti-hormone treatments. We will also interrogate the mechanistic contribution of histone modifications and other epigenetic modulators for the establishment of transition TAD structures. In silico expression profiling and single-cell RNA seq will be conducted in primary tumors of TCGA cohorts and in cancer cell subpopulations, respectively, and determine whether concordant regulation of TAD-associated target genes is intrinsic predictors of hormone resistance in the breast or prostate cancer.